
Mind the Matrix: Looking Beyond Old Cells in Brain Aging
The brain extracellular matrix - what is it and who cares?


A lot of things can go wrong with cells in the brain as we age, and the same is true for the environment that these cells exist in. Extracellular space accounts for a whopping 20% of the volume in the adult brain [1] and this space is filled with the extracellular matrix (ECM), a rich meshwork of proteins, sugars, and other macromolecules [2]. These molecules surround, influence, and are in turn shaped by the cells of the nervous system in highly dynamic and highly underappreciated ways.
Despite the considerable space it takes up, the brain ECM is often ignored. Researchers instead focus on the cells in the brain as if they existed in a vacuum. For the scientists out there, this is perhaps best illustrated with a thought experiment. Imagine you want to study your favorite cells in a dish, whether they are neurons or glia. How often do you consider culturing them on a halfway decent extracellular matrix, let alone any at all?
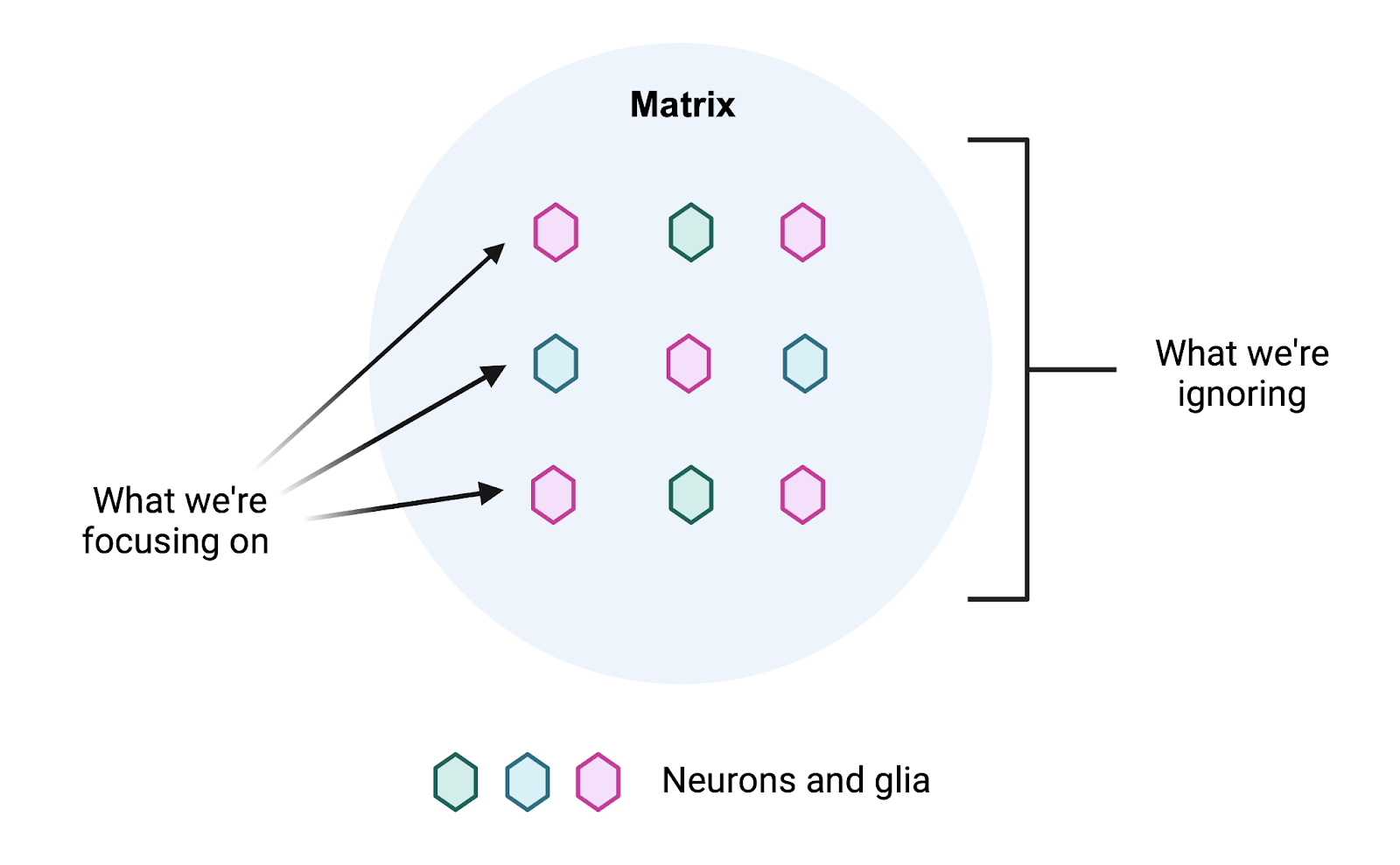
Ironically, “glia” is derived from the Greek word for glue, as such cells were mistakenly thought to serve as a connective structure in the brain [3]. This term would perhaps more aptly describe the ECM, although it does far more than simply provide structural support. In fact, the ECM interfaces with multiple pathways critical to brain aging. As a result, any attempts to intervene in this process will have to contend with the matrix.
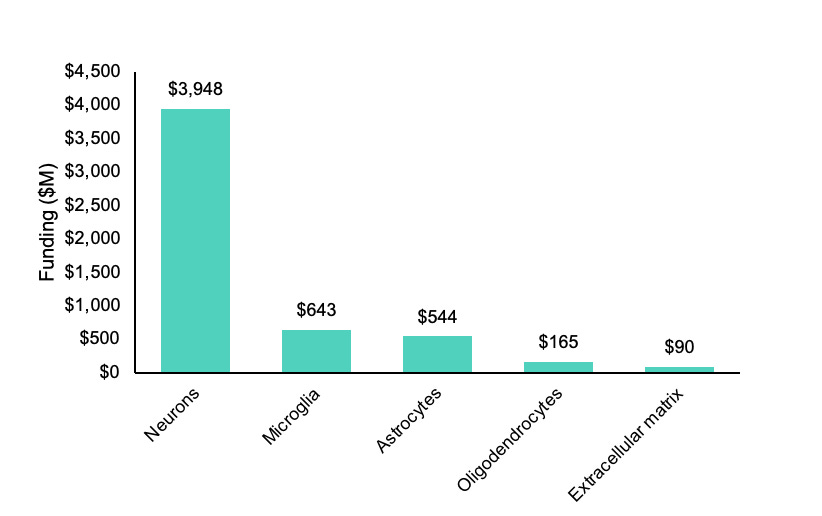
At the time of writing, there is only ~$90M in active funding combined from the two primary government sponsors of brain and aging research for projects containing the key words “extracellular matrix” in the National Institutes of Health’s RePORTER database. A crude measure to be sure, but comparing this to the ~$643M in “microglia” projects by these agencies reveals a pretty apparent lack of interest in the matrix relative to glial cells, let alone neurons.
Using the framework provided by age1 to classify aging interventions, I’ll illustrate how the ECM will be critical to three of the major approaches as they apply to the brain. These strategies include delaying brain aging and replacing or restoring aged cells and tissues. Then I’ll get into what we can do about it.
In some cases, the relevance of the ECM is intuitive - if you were going to replace your old cells with shiny new ones, would you want to put them in the same old, junk-filled environment?
In other cases, a deeper understanding of the brain ECM will be necessary to fully grasp its importance.
Delaying aging: The matrix in neuroplasticity
The brain extracellular matrix can be divided into three primary compartments:
The diffuse ECM, located in the interstitial space and around synapses
Perineuronal nets, dense ECM coats that form around certain neurons
The basement membrane of the blood-brain barrier
Each of these compartments has distinct structural and functional characteristics [4].
The diffuse ECM is a loose network of proteins and sugars that surrounds cells and synapses throughout the brain, where in the latter case it is referred to as the perisynaptic matrix. It’s distinct in its molecular makeup from ECM found in other parts of the body, reflecting its specialized role in brain tissue.
In contrast, perineuronal nets (PNNs) are more structurally compact and surround only certain neuronal subtypes. Although PNNs share many of the same matrix components as the diffuse ECM, these components differ in their concentration and organization.
The basement membrane, with its own unique repertoire of matrix molecules, lines the blood vessels of the brain and supports the integrity of the blood-brain barrier.
With this in mind, there are two major changes to the brain ECM that occur with age and associated pathologies that can be targeted to delay brain aging, and both of these directly impact synaptic plasticity:
A build-up of diffuse matrix material around synapses
A breakdown of perineuronal nets
Diffuse ECM builds up
Brain plasticity, a frequent target of therapeutic efforts, is critically regulated by the matrix. Matrix material builds up in the brain interstitial space with age [5], in the so-called diffuse ECM, where it accumulates around synapses and restricts synaptic plasticity [6]. Similar diffuse ECM builds up in neurodegeneration, as in Alzheimer’s [7] and Huntington’s disease [8], where deficits in synaptic plasticity are also rampant.
The maintenance of healthy synapses is critical to cognitive health in aging [9], and it may be possible to attenuate age-related synaptic deficits by reducing or preventing the accumulation of ECM around synapses. This can be done by directing the brain’s resident immune cells, microglia, to clear perisynaptic ECM via cytokine delivery, as has been shown with IL-33 [6]. Alternatively, enzymes could be delivered to break down matrix material directly [10]. Both approaches enhance synaptic plasticity.
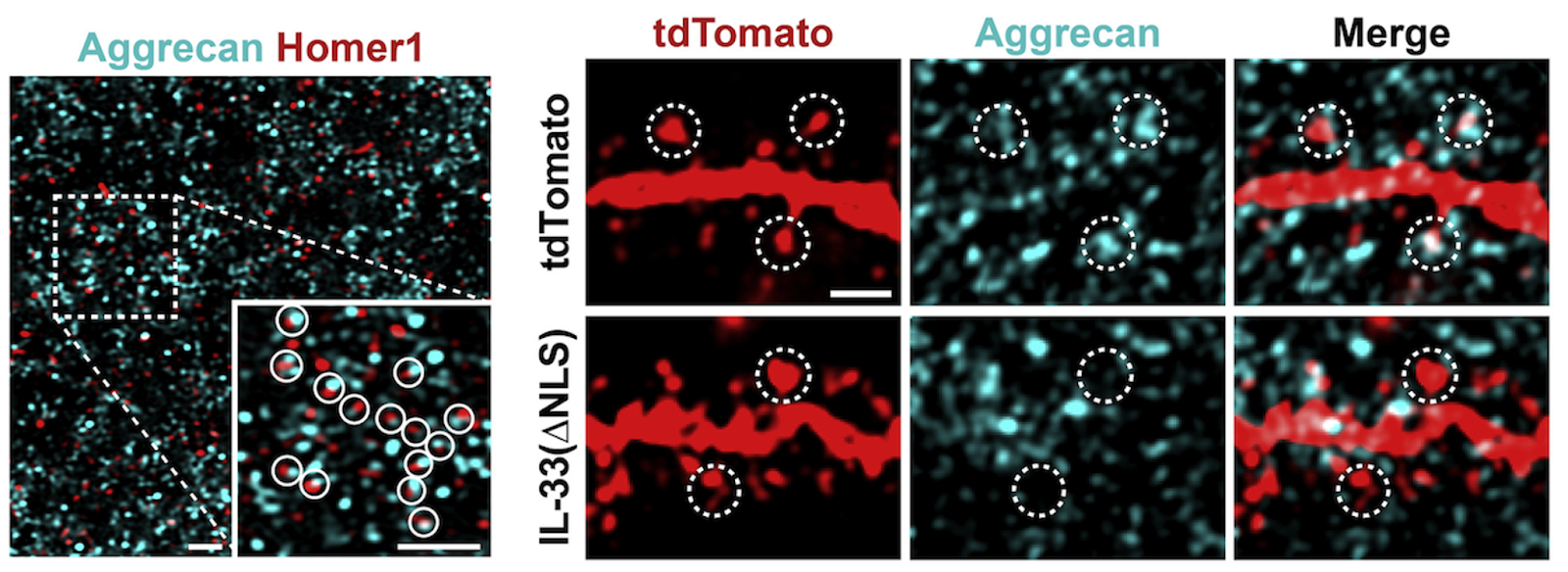
Perineuronal nets break down
At the same time that the diffuse interstitial matrix is building up with age, the ECM is breaking down in other parts of the brain. In contrast to the diffuse ECM, extracellular proteins and polysaccharides can also condense around specific subsets of neurons in a lattice-like structure known as the perineuronal net, which serves to stabilize and support synaptic architecture [11].
The formation of PNNs is a critical event that brings about the closure of heightened periods of plasticity in the developing brain. By stabilizing synapses in certain neurons, PNNs fundamentally control and restrict synaptic plasticity. In line with this, their removal in the adult brain is able to reinstate plasticity to an extent seen in early brain development [12, 13].
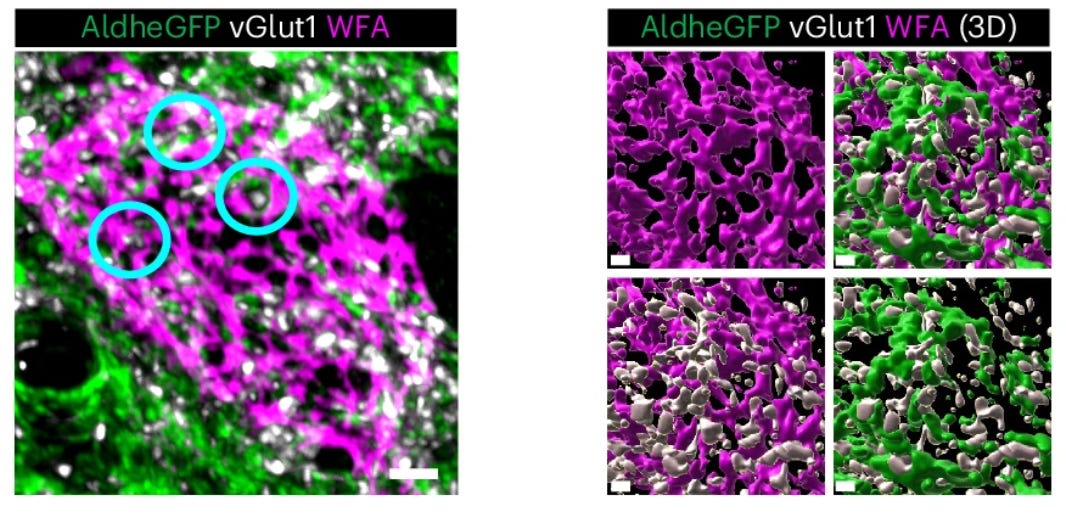
In my graduate work with Dr. Kim Green at UC Irvine, we found that PNNs are disrupted during the course of normal aging as well as in Alzheimer’s disease [7], and that this process is also regulated by microglia [14]. In Alzheimer’s disease patients, the loss of nets correlates with amyloid plaque load, and ECM components are found deposited in plaques [7].
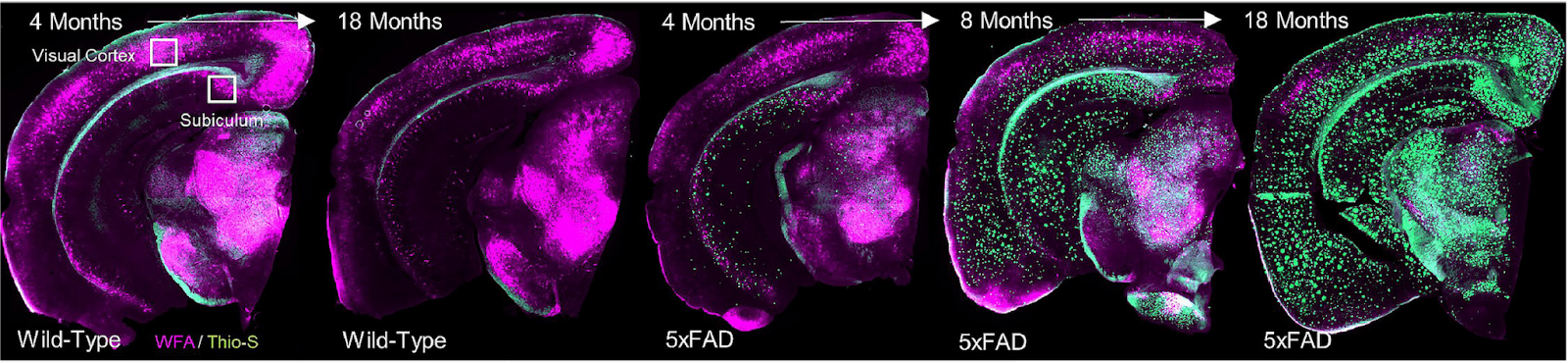
But the loss of PNNs would mean more synaptic plasticity, which is a good thing – right? It’s complicated. Particularly when it comes to a hallmark deficit of aging: memory.
Many subtypes of memory - from spatial to social to fear memories - are critically regulated by PNNs [15]. Take episodic memory, through which we remember our personal experiences as we go about our daily lives. The ability to form precise memories of events develops after birth, with young children only able to form imprecise, fuzzy memories until this system comes online at ~5-8 years of age [16].
In mice, this process is controlled by the maturation of hippocampal PNNs, and adult episodic-like memory precision can be produced by bringing about PNN formation in the brains of younger mice [16]. Memory imprecision characteristic of the immature brain occurs in adults if PNNs are removed.
The Nobel Prize-winning chemist Roger Tsien proposed that PNNs may serve as the physical basis for long-term memory storage [17]. While this is a pretty grandiose claim, it is clear that PNNs and memory are closely intertwined, albeit often in ways that remain somewhat opaque.
The increased plasticity required to encode new memories has to be balanced with sufficient synaptic stability to retain the old ones [11]. As such, PNN removal may enhance plasticity at the cost of long-term memory stability, due to interference with new memories [18]. So by disrupting PNNs, we might be sacrificing the fidelity of our memories for this boost in plasticity.
For more information on PNNs in aging and AD, and their role in plasticity/memory, see:
A review on the role of microglia in ECM and PNN regulation by yours truly, which more thoroughly covers the literature on ECM changes in aging and neurodegeneration
A primer on PNNs in memory by Fawcett et al., a leading PNN expert
A review by Reichelt et al. on PNNs in plasticity and the “stability-plasticity tradeoff”
A dual approach: Compartment-specific matrix regulation for brain health
In order to delay brain aging, efforts may be best aimed at reducing the build-up of perisynaptic ECM while simultaneously preventing the breakdown or loss of perineuronal nets. With this approach, we could promote general synaptic plasticity and integrity while still maintaining the stability of long-term memory storage.
As a bonus, PNNs are inherently protective against age-related pathologies, protecting the neurons they enwrap from toxicity associated with amyloid beta [19], oxidative stress [20], and tau [21]. They also keep neurotransmitters from spilling over outside of their synapses [22]. It’s not a stretch to imagine then that PNN maintenance could provide protection for the aging brain across multiple dimensions.
Replacing the aging brain: The matrix in cell transplantation approaches
Replacing cells
Beyond plasticity, the brain ECM actively regulates functions of local brain cells including survival, proliferation, differentiation and migration [4]. Approaches to replace aging or diseased cells in the brain via transplantation of healthy cells will have to factor in the ECM, which accumulates its own age-related damage.
Aged ECM is stiffer, for instance, at least in the rat brain (things are less clear in humans - see the Appendix). This mechanical change impairs the ability of oligodendrocyte precursor cells to proliferate and differentiate. In line with this, precursor cells from neonatal rats lose their proliferative capabilities when seeded on aged ECM [23].
Putting new cells into the brain without considering the extracellular environment in which they exist would be like trying to put a human on Mars without planning for the unique atmosphere, and yet transplantation efforts seldom consider it.
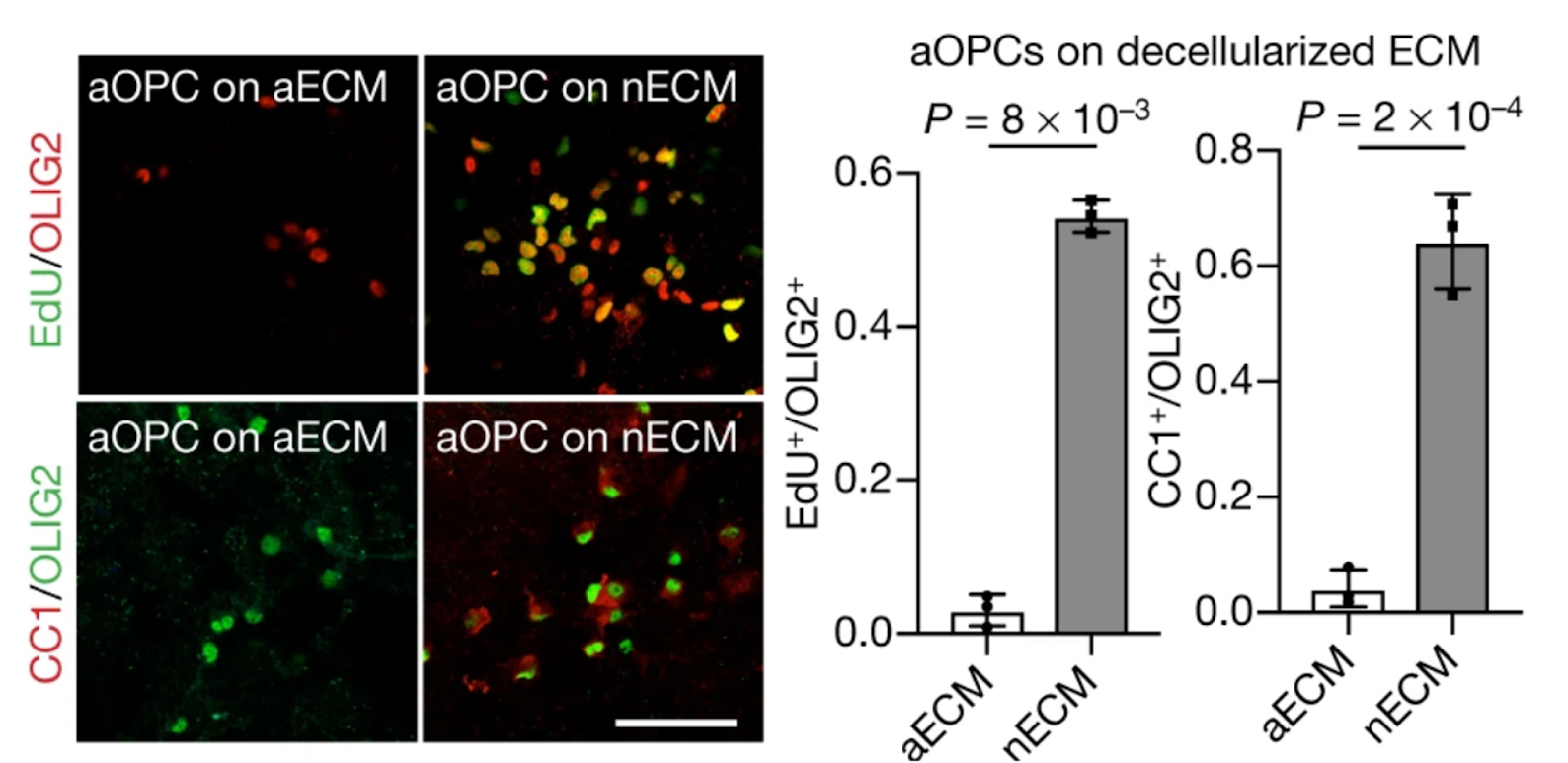
Brain cell replacement therapies should therefore develop and incorporate auxiliary strategies to make the aged ECM more hospitable. One potential approach is to deliver enzymes that temporarily break down specific ECM components alongside transplanted cells. This method could temporarily modify tissue stiffness and reduce synaptic ECM accumulation, facilitating integration of donor cells and promoting therapeutic efficacy.
Alternatively, transplanted cells could be engineered to express their own ECM-degrading enzymes, or a unique repertoire of integrins, which are receptors by which cells bind to and interact with the matrix. These could be fine-tuned to maximize integration and function of transplanted cells, much like the spacesuit we would give our astronaut to ensure survival on another planet.
Replacing ECM?
It would be interesting to consider whether replacement of the ECM itself could serve as a viable therapeutic strategy to combat brain aging. Could old, beaten-up ECM that likely carries similar age-related damage as reported for the matrix in other tissues [24] be broken down and replaced by the delivery of young ECM? Certainly for the diffuse matrix this seems possible.
In PNNs, on the other hand, matrix components are tightly packed together by what are known as link proteins. Overexpression of these link proteins can induce PNN formation where PNNs are lacking [16]. One possibility might be to deliver young ECM around neurons with disrupted PNNs and then promote link protein expression, with the hope that the donor ECM would be incorporated into new nets.
Given the importance of the ECM in cellular health, plasticity, and synaptic function, these replacement strategies could be undertaken as primary therapies or as adjuncts to promote the efficacy of cell transplantations.
Restoring a youthful brain: The matrix in neurogenesis
Strategies to counter brain aging may also look to exploit natural mechanisms that promote neuronal rejuvenation and that already exist in nervous tissue, as in neurogenesis. Here, too, we have to contend with the matrix.
Regions of the adult brain where new neurons are formed - neurogenic niches - have their own specialized ECM that is stiffer than non-neurogenic regions [25]. Further age-related alterations in niche stiffness might directly impact neurogenesis. In fact, neural stem cells are more adhesive and have difficulty migrating out of certain neurogenic niches with age [26], a deficit that may be impacted by local changes in ECM stiffness.
By targeting the ECM, we may be able to create environments that better support the brain’s own repair mechanisms, potentially slowing or even reversing age-related decline.
Interestingly, the enzymatic removal of ECM in neurogenic niches can impair neurogenesis [27]. This might be related to the ability of the ECM to serve as a reservoir for important cellular growth factors. Increasing [28] or decreasing [29] specific ECM components can also boost or inhibit neurogenesis, respectively. A key challenge in developing therapies will be to selectively target the detrimental age-related accumulation of ECM without impairing the essential pro-neurogenic functions of the healthy, homeostatic matrix - and ideally, even enhancing them.
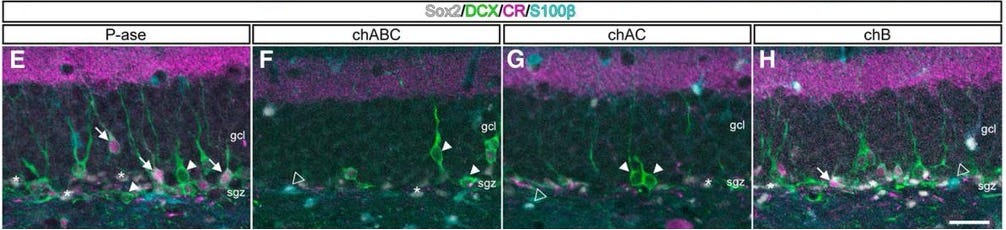
There’s far more to talk about on the role of the ECM in neurogenesis than we have room for here, and work is still being done. But it’s clear that there’s a lot we can do to influence these endogenous restorative pathways in aging by tinkering with the matrix.
For more information:
A review on the ECM-neurogenesis interface by Elise Cope and Elizabeth Gould
A discussion on the matrix in the neurogenic niche in a review by Dityatev et al.
Okay, the ECM is important in aging. So what can we do about it?
We can:
Look into the matrix
Manipulate the matrix
Looking into the matrix
In a proteomic study of blood plasma from over 40,000 individuals, higher levels of specific brain-derived ECM proteins were associated with a more youthful brain age. Even more striking, perineuronal nets and synapse-associated ECM were the top two brain protein pathways associated with human longevity [30].
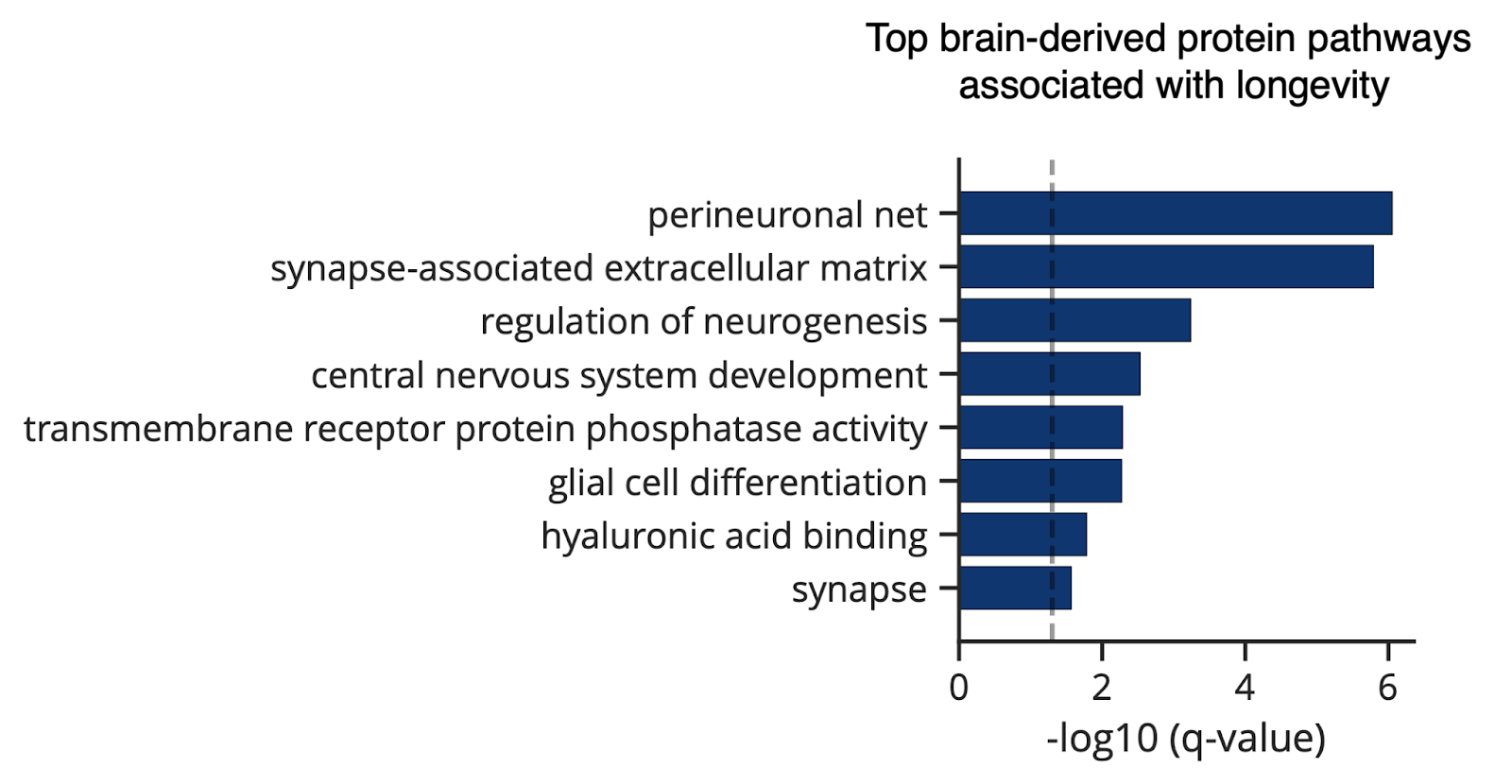
By characterizing the brain ECM and how it changes with age and age-related disease, we can determine the best way to factor it into brain aging interventions.
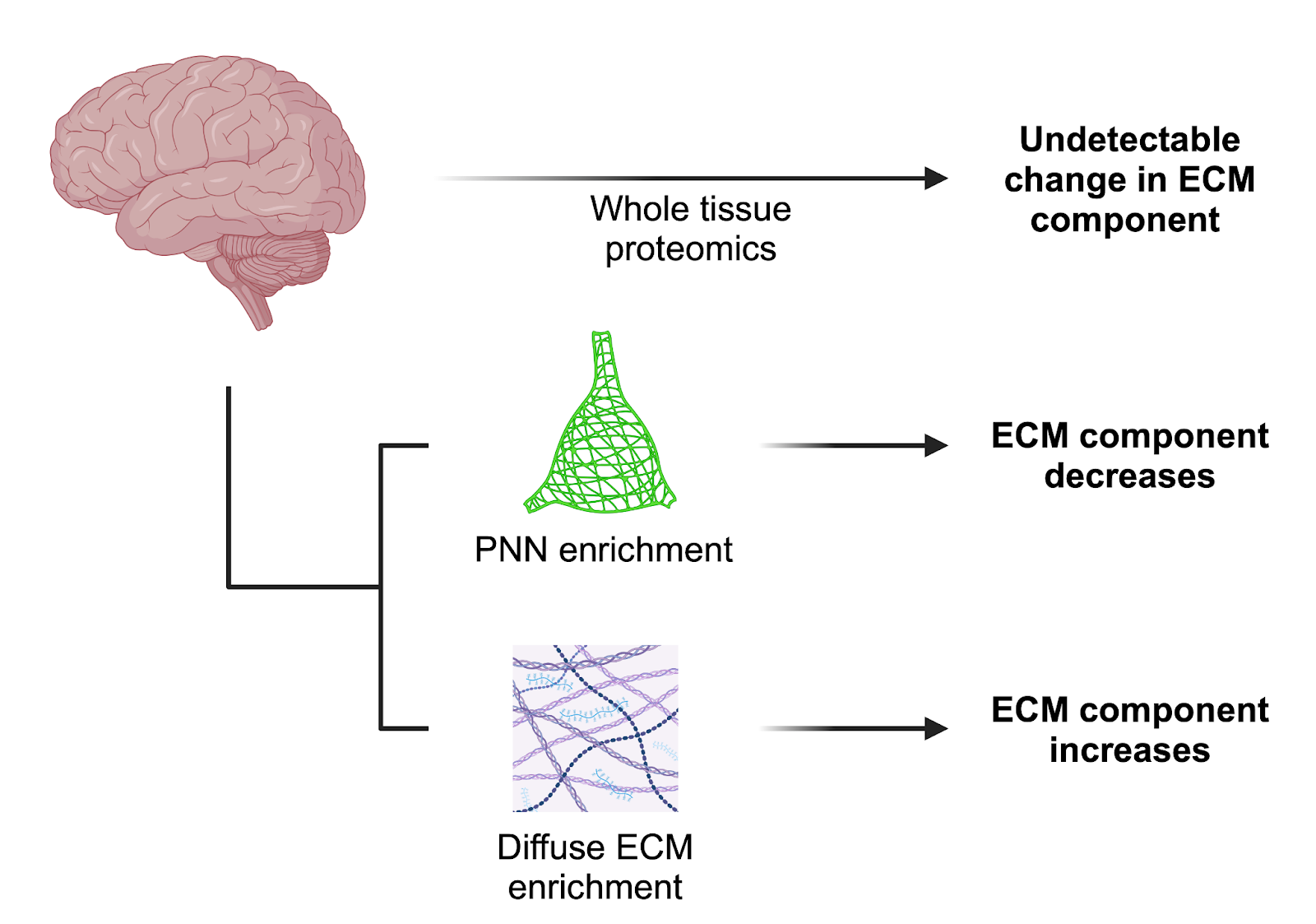
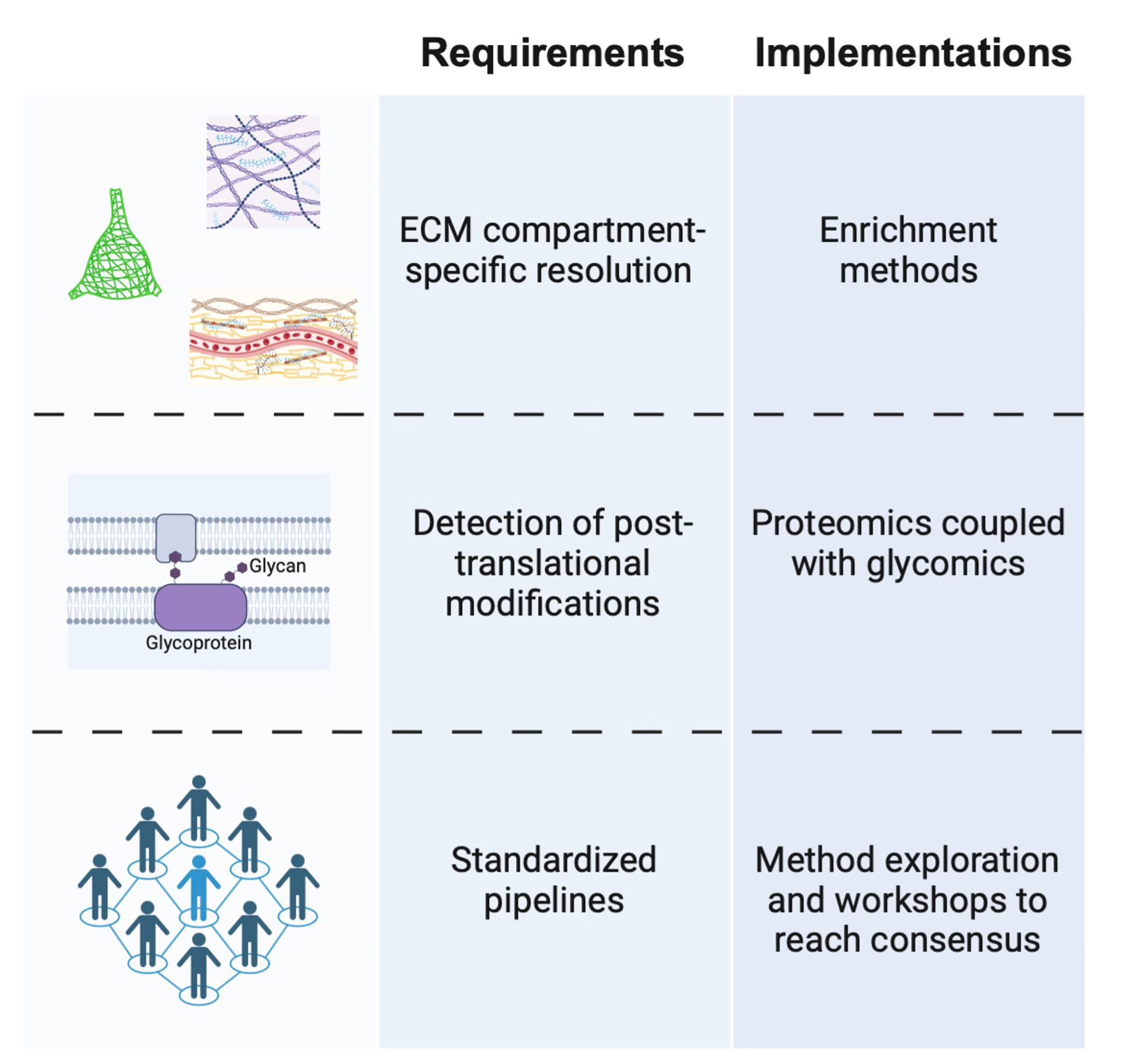
There remain gaps in our understanding of the precise compositional differences between the ECM compartments of the brain, as well as how these differences are regulated. Proteomic screens to define the ‘matrisome’ [31] have been and will continue to be critical, with a strong emphasis on human data. While transcriptomic analysis is important, the secretory and long-lived nature of ECM components to some extent uncouples them from transcriptional dynamics.
One particular challenge is that the different brain ECM compartments - PNNs, diffuse ECM, and basement membranes - overlap in the proteins and sugars they contain. Changes at these smaller scales may be missed with proteomic analysis of bulk tissue. Enrichment approaches have been developed to address this issue, as in the isolation of cerebrovascular- [32] and PNN-associated [33] matrices, and should be applied to studies on the aging brain.
To further complicate things, ECM molecules are heavily post-translationally modified, and these modifications impart critical functional properties [34]. This often occurs through the attachment of sugar side chains that, in turn, have their own age-related chemical modifications. Combining proteomics with glycomics to capture these modifications will be most fruitful [35].
At the moment, different approaches are taken by researchers to characterize the various ECM compartments and components, making integration and comparison of data across studies difficult. Ultimately, a consensus will need to be reached in the brain ECM community about the methodology most suitable for screening the matrix and its changes so that we know exactly what we’re trying to fix.
To really understand the biology of matrix constituents, these approaches need to be complemented with thorough functional experimentation, and our ability to do so depends on the methodological tools at hand. However, the availability of tools to manipulate the brain ECM in vivo is fundamentally limited, and progress in studying and consequently targeting the brain ECM therapeutically will depend on addressing this roadblock.
Manipulating the matrix
The most commonly used method in studying matrix function is to take the molecular equivalent of a sledgehammer to it and see how this affects your favorite biological process. Chondroitinase ABC, a bacterial enzyme that cleaves the side chains of certain brain ECM-enriched molecules, has been delivered to nervous tissue in vivo as an investigational technique for over twenty years [36].
As someone who spent his time in graduate school studying microglia and their influence on the ECM by pharmacologically removing them from the brain, I’m the first to acknowledge the utility of such heavy-handed approaches in revealing fundamental biology [37].
However, chondroitinase has many drawbacks:
It generally requires direct, invasive delivery to the brain
Because it quickly loses activity at 37°C, ECM effects are temporary
It’s nonspecific, acting on a family of ECM molecules (CSPGs)
At the same time, it only acts on one subset of ECM molecules
The degradation products it generates from cleaving ECM can directly influence inflammation [38, 39] and confound experiments via off-target effects
This tool is a mainstay of ECM research and is constantly being upgraded. However, we need to acknowledge its limitations. Enzymes that act on other matrix components [40, 41] can also be used but may run into similar problems related to invasiveness of delivery and the generation of byproducts with their own biological activities, resulting in off-target effects [42].
So what else is there? Mice with ECM-related genes knocked out are a good bet. A number of mouse lines have been created that lack expression of individual ECM molecules [43-46] or the enzymes that synthesize [47] or modify them [34]. More advanced genetic models are now available that enable conditional manipulation of some of these proteins in particular cell types [13].
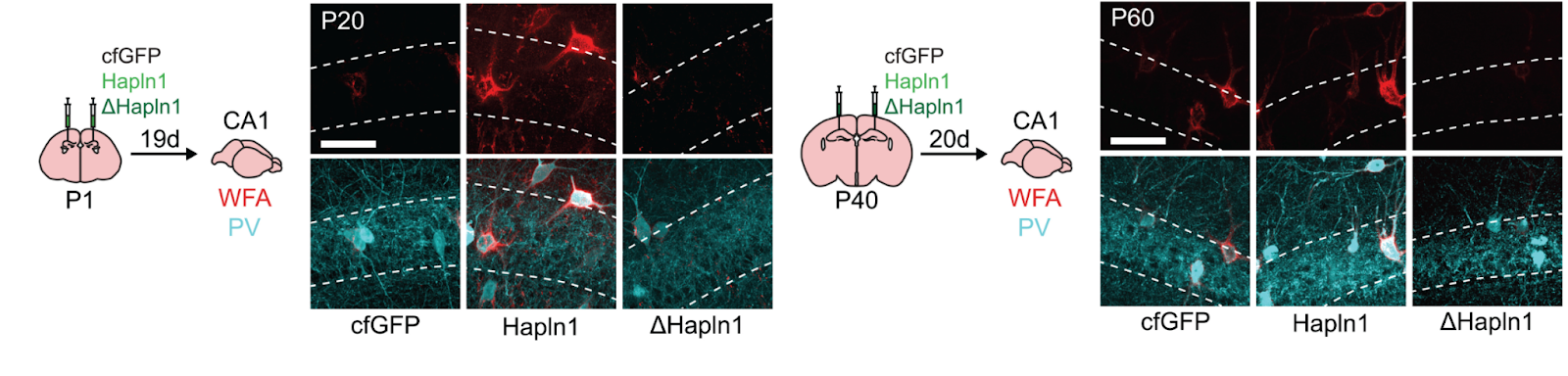
ECM substructure specificity is also possible, to a degree, as indicated earlier: by genetically removing the link proteins that hold the components of PNNs together, you can disrupt PNNs with minimal effects on the total levels of other core ECM components [48, 49]. Viruses driving the expression of a link protein or its mutant variant can be delivered to promote PNN formation and disassembly, respectively [16].
To the point made in the beginning of this article about in vitro experiments often neglecting the ECM: culturing methods have been developed to grow neurons and glia on decellularized brain tissue [23, 50], so the ECM can be manipulated both in an organism and in a dish. The development of ex vivo post-mortem brain perfusion systems [51] will further advance the translational potential of ECM screening approaches and functional experimentation.
Existing approaches to manipulating the brain ECM:
Chondroitinase and other ECM-cleaving enzymes
Constitutive and inducible cell type-specific ECM knockout mice or tissue
Mouse lines lacking enzymes that synthesize or chemically modify the ECM
Viral delivery of link protein HAPLN1 or a mutant variant to induce PNN formation or loss
In vitro systems using decellularized brain tissue
A major roadblock in the field is the lack of small molecules that can regulate ECM synthesis, modification, and clearance. This relegates experimental studies to genetic, viral, or enzymatic approaches, which, while useful, are cumbersome and/or often nonspecific. And they are much more difficult to get off the ground translationally.
The tools we need for the human matrix
What we really need are better pharmacological agents that can act on the ECM, either directly or indirectly.
Important examples currently in use include:
A peptide that interrupts a specific ECM-receptor interaction [52]
An antibody that binds to and blocks an inhibitory ECM component [53]
Several small molecules that act as substrate analogues in ECM synthesis, which can reduce levels of core ECM proteins and the elongation of their sugar side chains [54, 55].
We need more of these. Ideally, we need small molecules that are ECM compartment-specific and that can shape the diffuse ECM, perineuronal nets, and/or vascular basement membrane ECM, either individually or in combination.
The development of blood-brain barrier-penetrant small molecules that activate or inhibit enzymes controlling ECM synthesis or degradation would unlock the potential for precise control over the ECM to an extent that is not currently possible. These could include drug candidates that act on proteolytic enzymes [56] or the molecules that regulate them [5], or compounds that target the enzymes directly involved in ECM synthesis and chemical modification.
Alternatively, one could hijack the brain’s natural ECM clean-up crew by targeting the cytokines that control microglial processing of the ECM, as with IL-33 [6], or even microglial activation more broadly as a condition associated with ECM remodeling [7]. Whatever the approach, we need better tools to manipulate the brain matrix that can be more readily applied to the clinic.
Conclusion
Returning to the question I asked in the beginning, who cares about the brain ECM? The sad news is not too many people, at least as far as funding is concerned. The graph of active research spend shown at the start was meant to be as inclusive as possible for projects related to the matrix on RePORTER by the National Institute on Aging and the National Institute of Neurological Disorders and Stroke, but funding from the former likely also includes projects related to body-wide ECM.
Searching for active projects specifically related to the brain extracellular matrix returned just ~$62M in ongoing funding. This needs to change.
We have only very briefly covered the biology of the brain ECM in aging and age-related disorders here. The hope is that those working on interventions for brain aging will reconsider the role of the ECM in their approach - but I’ll settle for it being considered at all. Researchers who may want to look into the matrix further in their own research now have a place to begin, and an idea of the problems that need to be addressed for therapeutic advancement.
Until we face the matrix, the possibility of restoring the aged brain may remain fundamentally out of reach.
Appendix
NIH funding breakdown
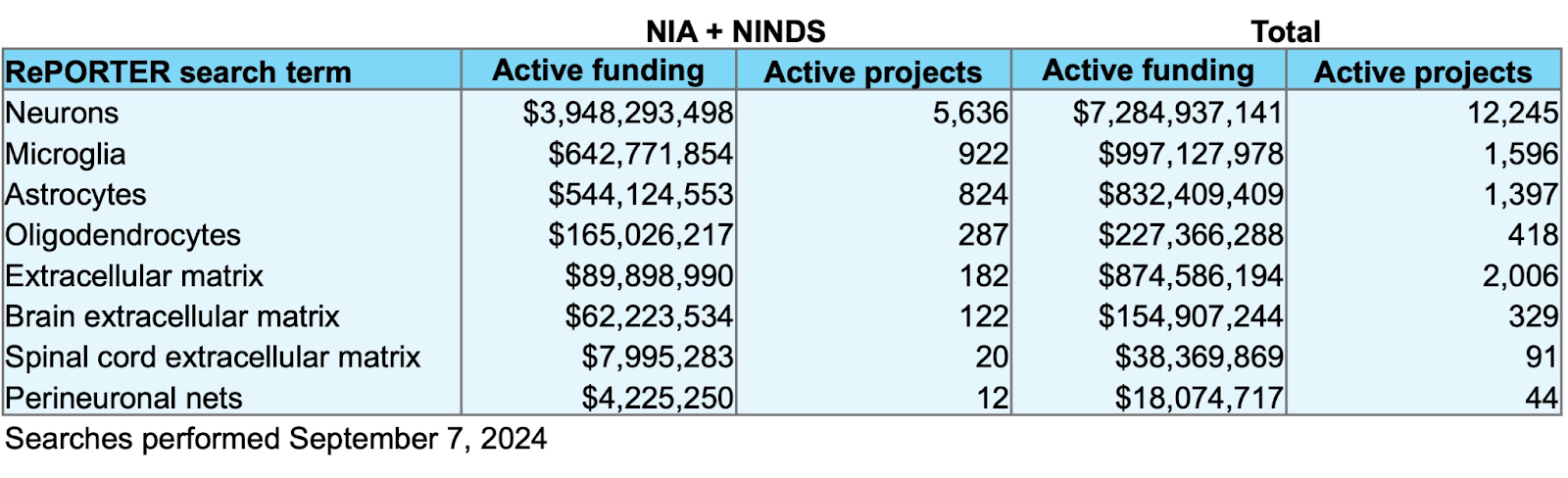
Data from RePORTER used to generate the funding chart. The funding is broken down into combined active funding by the National Institute on Aging and the National Institute of Neurological Disorders and Stroke as well as the total funding reported across all institutes and agencies. The total numbers of active projects that are currently funded are also displayed.
It’s interesting to note that there is a sizable investment in the extracellular matrix outside of the brain, with ~$875M total funding across institutes. Over a third of this comes from active projects sponsored by the National Heart, Lung, and Blood Institute (~$225M) and the National Cancer Institute (~$120M). Researchers studying the brain ECM may therefore benefit from the insights gained in these fields, such as the finding that local ECM stiffness hinders therapeutic delivery and immune cell infiltration into tumors [57]. Similar factors may affect therapeutic delivery to the aging brain.
Still, the numbers for total ECM funding - accounting for the matrix across all organs and tissues - barely hold up to the funding directed to individual glial cell types.
The search conditions used to generate these numbers are found in the table below. Searches were intended to be as inclusive as possible while still retaining sufficient specificity.

Additional notes on the aging brain matrix
There are other aspects of brain ECM biology that are implicated in aging but that lie outside of the scope of this article:
ECM constituents are chemically modified during aging in a way that contributes to plasticity and memory impairments [34]
ECM molecules deposit in amyloid plaques [58, 59] and clearing ECM molecules can reduce amyloid plaque pathology [40, 58, 59]
Genetic variants that can stave off cognitive decline in familial Alzheimer’s disease patients by decades are related to their effects on ECM molecules [60]
In addition to being regulated by immune cells, the ECM directly regulates inflammation and serves as a reservoir of immune-relevant cytokines
Interestingly, whereas the rat brain stiffens, human brains display an overall softening with age [61]. It is unclear the extent to which this is due to cell loss vs. changes in the mechanical properties of the ECM itself.
It may be the case that ECM in the human brain stiffens during the course of aging, as in the aging rat brain, while age-related cell loss and atrophy leads to a general softening of the human brain overall. Amyloid fibrils that accumulate in the brain extracellular space in Alzheimer’s disease are exceptionally stiff [62], for instance, and may augment the stiffness of the local ECM while the disease-related loss of cells and synapses in humans results in softening at the macro scale.
Further research is needed to close this translational gap. In any case, given what we know about the effects of the ECM on the functions of endogenous and transplanted cells - which extend beyond just its mechanical properties - it will remain prudent to keep the matrix in mind when developing strategies to treat brain aging.
Literally looking into the matrix
Recent developments in live imaging over the last year have enabled the use of dyes and fluorescently-labeled compounds to visualize the major brain ECM subcompartments in vivo in real-time [63, 64]. Live imaging of brain ECM and PNNs in vivo is also now possible with the use of a fluorescent probe that is genetically fused to an important ECM protein (the link protein HAPLN1) [65], and the mice are available on JAX. Another fusion-labeling system based on this same protein has already been developed to both birthdate brain ECM and to reveal novel matrix architecture [66]. Applying these tools to the aging brain will allow us to see how the ECM changes at a resolution never before possible.
Acknowledgements
As stated earlier, the framework used to address the importance of the ECM in brain aging was inspired by and based on age1’s categories of interventions for aging. Inspiration was also drawn from our team’s own Bottlenecks of Aging and research roadmap for combatting brain aging. Infographics were generated with BioRender unless otherwise indicated.
Huge thanks to everyone at the Amaranth Foundation for helping this come together - Joanne Peng for the constant feedback, Jarod Rutledge, Rohan Krajeski, and Raiany Romanni (several times over) for their thoughts and comments, and Alex Colville for input and for suggesting I do this in the first place. And of course thank you to James Fickel for making this possible.
Nicholson, C., and Syková, E. (1998). Extracellular space structure revealed by diffusion analysis. Trends Neurosci. 21, 207–215. https://doi.org/10.1016/S0166-2236(98)01261-2.
Dityatev, A., Seidenbecher, C.I., and Schachner, M. (2010). Compartmentalization from the outside: the extracellular matrix and functional microdomains in the brain. Trends Neurosci. 33, 503–512. https://doi.org/10.1016/j.tins.2010.08.003.
Allen, N.J., and Lyons, D.A. (2018). Glia as Architects of Central Nervous System Formation and Function. Science 362, 181–185. https://doi.org/10.1126/science.aat0473.
Lau, L.W., Cua, R., Keough, M.B., Haylock-Jacobs, S., and Yong, V.W. (2013). Pathophysiology of the brain extracellular matrix: a new target for remyelination. Nat. Rev. Neurosci. 14, 722–729. https://doi.org/10.1038/nrn3550.
Ferreira, A.C., Hemmer, B.M., Philippi, S.M., Grau-Perales, A.B., Rosenstadt, J.L., Liu, H., Zhu, J.D., Kareva, T., Ahfeldt, T., Varghese, M., et al. (2023). Neuronal TIMP2 regulates hippocampus-dependent plasticity and extracellular matrix complexity. Mol. Psychiatry 28, 3943–3954. https://doi.org/10.1038/s41380-023-02296-5.
Nguyen, P.T., Dorman, L.C., Pan, S., Vainchtein, I.D., Han, R.T., Nakao-Inoue, H., Taloma, S.E., Barron, J.J., Molofsky, A.B., Kheirbek, M.A., et al. (2020). Microglial Remodeling of the Extracellular Matrix Promotes Synapse Plasticity. Cell 182, 388-403.e15. https://doi.org/10.1016/j.cell.2020.05.050.
Crapser, J.D., Spangenberg, E.E., Barahona, R.A., Arreola, M.A., Hohsfield, L.A., and Green, K.N. (2020). Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58, 102919. https://doi.org/10.1016/j.ebiom.2020.102919.
Crapser, J.D., Ochaba, J., Soni, N., Reidling, J.C., Thompson, L.M., and Green, K.N. (2020). Microglial depletion prevents extracellular matrix changes and striatal volume reduction in a model of Huntington’s disease. Brain 143, 266–288. https://doi.org/10.1093/brain/awz363.
Morrison, J.H., and Baxter, M.G. (2012). The Aging Cortical Synapse: Hallmarks and Implications for Cognitive Decline. Nat. Rev. Neurosci. 13, 240–250. https://doi.org/10.1038/nrn3200.
Orlando, C., Ster, J., Gerber, U., Fawcett, J.W., and Raineteau, O. (2012). Perisynaptic Chondroitin Sulfate Proteoglycans Restrict Structural Plasticity in an Integrin-Dependent Manner. J. Neurosci. 32, 18009–18017. https://doi.org/10.1523/JNEUROSCI.2406-12.2012.
Reichelt, A.C., Hare, D.J., Bussey, T.J., and Saksida, L.M. (2019). Perineuronal Nets: Plasticity, Protection, and Therapeutic Potential. Trends Neurosci. 42, 458–470. https://doi.org/10.1016/j.tins.2019.04.003.
Pizzorusso, T., Medini, P., Berardi, N., Chierzi, S., Fawcett, J.W., and Maffei, L. (2002). Reactivation of Ocular Dominance Plasticity in the Adult Visual Cortex. Science 298, 1248–1251. https://doi.org/10.1126/science.1072699.
Rowlands, D., Lensjø, K.K., Dinh, T., Yang, S., Andrews, M.R., Hafting, T., Fyhn, M., Fawcett, J.W., and Dick, G. (2018). Aggrecan Directs Extracellular Matrix-Mediated Neuronal Plasticity. J. Neurosci. 38, 10102–10113. https://doi.org/10.1523/JNEUROSCI.1122-18.2018.
Crapser, J.D., Arreola, M.A., Tsourmas, K.I., and Green, K.N. (2021). Microglia as hackers of the matrix: sculpting synapses and the extracellular space. Cell. Mol. Immunol. 18, 2472–2488. https://doi.org/10.1038/s41423-021-00751-3.
Fawcett, J.W., Fyhn, M., Jendelova, P., Kwok, J.C.F., Ruzicka, J., and Sorg, B.A. (2022). The extracellular matrix and perineuronal nets in memory. Mol. Psychiatry 27, 3192–3203. https://doi.org/10.1038/s41380-022-01634-3.
Ramsaran, A.I., Wang, Y., Golbabaei, A., Aleshin, S., de Snoo, M.L., Yeung, B.A., Rashid, A.J., Awasthi, A., Lau, J., Tran, L.M., et al. (2023). A shift in the mechanisms controlling hippocampal engram formation during brain maturation. Science 380, 543–551. https://doi.org/10.1126/science.ade6530.
Tsien, R.Y. (2013). Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc. Natl. Acad. Sci. 110, 12456–12461. https://doi.org/10.1073/pnas.1310158110.
Christensen, A.C., Lensjø, K.K., Lepperød, M.E., Dragly, S.-A., Sutterud, H., Blackstad, J.S., Fyhn, M., and Hafting, T. (2021). Perineuronal nets stabilize the grid cell network. Nat. Commun. 12, 253. https://doi.org/10.1038/s41467-020-20241-w.
Miyata, S., Nishimura, Y., and Nakashima, T. (2007). Perineuronal nets protect against amyloid beta-protein neurotoxicity in cultured cortical neurons. Brain Res. 1150, 200–206. https://doi.org/10.1016/j.brainres.2007.02.066.
Cabungcal, J.-H., Steullet, P., Morishita, H., Kraftsik, R., Cuenod, M., Hensch, T.K., and Do, K.Q. (2013). Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 110, 9130–9135. https://doi.org/10.1073/pnas.1300454110.
Suttkus, A., Holzer, M., Morawski, M., and Arendt, T. (2016). The neuronal extracellular matrix restricts distribution and internalization of aggregated Tau-protein. Neuroscience 313, 225–235. https://doi.org/10.1016/j.neuroscience.2015.11.040.
Tewari, B.P., Woo, A.M., Prim, C.E., Chaunsali, L., Patel, D.C., Kimbrough, I.F., Engel, K., Browning, J.L., Campbell, S.L., and Sontheimer, H. (2024). Astrocytes require perineuronal nets to maintain synaptic homeostasis in mice. Nat. Neurosci., 1–14. https://doi.org/10.1038/s41593-024-01714-3.
Segel, M., Neumann, B., Hill, M.F., Weber, I., Viscomi, C., Zhao, C., Young, A., Agley, C.C., Thompson, A.J., Gonzalez, G., et al. (2019). Niche stiffness underlies the aging of central nervous system progenitor cells. Nature 573, 130–134. https://doi.org/10.1038/s41586-019-1484-9.
Fournet, M., Bonté, F., and Desmoulière, A. (2018). Glycation Damage: A Possible Hub for Major Pathophysiological Disorders and Aging. Aging Dis. 9, 880–900. https://doi.org/10.14336/AD.2017.1121.
Kjell, J., Fischer-Sternjak, J., Thompson, A.J., Friess, C., Sticco, M.J., Salinas, F., Cox, J., Martinelli, D.C., Ninkovic, J., Franze, K., et al. (2020). Defining the Adult Neural Stem Cell Niche Proteome Identifies Key Regulators of Adult Neurogenesis. Cell Stem Cell 26, 277-293.e8. https://doi.org/10.1016/j.stem.2020.01.002.
Yeo, R.W., Zhou, O.Y., Zhong, B.L., Sun, E.D., Navarro Negredo, P., Nair, S., Sharmin, M., Ruetz, T.J., Wilson, M., Kundaje, A., et al. (2023). Chromatin accessibility dynamics of neurogenic niche cells reveal defects in neural stem cell adhesion and migration during aging. Nat. Aging 3, 866–893. https://doi.org/10.1038/s43587-023-00449-3.
Yamada, J., Nadanaka, S., Kitagawa, H., Takeuchi, K., and Jinno, S. (2018). Increased Synthesis of Chondroitin Sulfate Proteoglycan Promotes Adult Hippocampal Neurogenesis in Response to Enriched Environment. J. Neurosci. 38, 8496–8513. https://doi.org/10.1523/JNEUROSCI.0632-18.2018.
Pujadas, L., Gruart, A., Bosch, C., Delgado, L., Teixeira, C.M., Rossi, D., Lecea, L. de, Martínez, A., Delgado-García, J.M., and Soriano, E. (2010). Reelin Regulates Postnatal Neurogenesis and Enhances Spine Hypertrophy and Long-Term Potentiation. J. Neurosci. 30, 4636–4649. https://doi.org/10.1523/JNEUROSCI.5284-09.2010.
Won, S.J., Kim, S.H., Xie, L., Wang, Y., Mao, X.O., Jin, K., and Greenberg, D.A. (2006). Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exp. Neurol. 198, 250–259. https://doi.org/10.1016/j.expneurol.2005.12.008.
Oh, H.S.-H., Le Guen, Y., Rappoport, N., Urey, D.Y., Rutledge, J., Brunet, A., Greicius, M.D., and Wyss-Coray, T. (2024). Plasma proteomics in the UK Biobank reveals youthful brains and immune systems promote healthspan and longevity. Preprint, https://doi.org/10.1101/2024.06.07.597771 https://doi.org/10.1101/2024.06.07.597771.
Naba, A., Clauser, K.R., Hoersch, S., Liu, H., Carr, S.A., and Hynes, R.O. (2012). The Matrisome: In Silico Definition and In Vivo Characterization by Proteomics of Normal and Tumor Extracellular Matrices. Mol. Cell. Proteomics MCP 11, M111.014647. https://doi.org/10.1074/mcp.M111.014647.
Pokhilko, A., Brezzo, G., Handunnetthi, L., Heilig, R., Lennon, R., Smith, C., Allan, S.M., Granata, A., Sinha, S., Wang, T., et al. (2021). Global proteomic analysis of extracellular matrix in mouse and human brain highlights relevance to cerebrovascular disease. J. Cereb. Blood Flow Metab. 41, 2423–2438. https://doi.org/10.1177/0271678X211004307.
Deepa, S.S., Carulli, D., Galtrey, C., Rhodes, K., Fukuda, J., Mikami, T., Sugahara, K., and Fawcett, J.W. (2006). Composition of perineuronal net extracellular matrix in rat brain: a different disaccharide composition for the net-associated proteoglycans. J. Biol. Chem. 281, 17789–17800. https://doi.org/10.1074/jbc.M600544200.
Yang, S., Gigout, S., Molinaro, A., Naito-Matsui, Y., Hilton, S., Foscarin, S., Nieuwenhuis, B., Tan, C.L., Verhaagen, J., Pizzorusso, T., et al. (2021). Chondroitin 6-sulphate is required for neuroplasticity and memory in ageing. Mol. Psychiatry 26, 5658–5668. https://doi.org/10.1038/s41380-021-01208-9.
Raghunathan, R., Hogan, J.D., Labadorf, A., Myers, R.H., and Zaia, J. (2020). A glycomics and proteomics study of aging and Parkinson’s disease in human brain. Sci. Rep. 10, 12804. https://doi.org/10.1038/s41598-020-69480-3.
Silver, J., and Miller, J.H. (2004). Regeneration beyond the glial scar. Nat. Rev. Neurosci. 5, 146–156. https://doi.org/10.1038/nrn1326.
Green, K.N., Crapser, J.D., and Hohsfield, L.A. (2020). To Kill a Microglia: A Case for CSF1R Inhibitors. Trends Immunol. 41, 771–784. https://doi.org/10.1016/j.it.2020.07.001.
Ebert, S., Schoeberl, T., Walczak, Y., Stoecker, K., Stempfl, T., Moehle, C., Weber, B.H.F., and Langmann, T. (2008). Chondroitin sulfate disaccharide stimulates microglia to adopt a novel regulatory phenotype. J. Leukoc. Biol. 84, 736–740. https://doi.org/10.1189/jlb.0208138.
Rolls, A., Cahalon, L., Bakalash, S., Avidan, H., Lider, O., and Schwartz, M. (2006). A sulfated disaccharide derived from chondroitin sulfate proteoglycan protects against inflammation-associated neurodegeneration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 20, 547–549. https://doi.org/10.1096/fj.05-4540fje.
Jendresen, C.B., Cui, H., Zhang, X., Vlodavsky, I., Nilsson, L.N.G., and Li, J.-P. (2015). Overexpression of Heparanase Lowers the Amyloid Burden in Amyloid-β Precursor Protein Transgenic Mice. J. Biol. Chem. 290, 5053–5064. https://doi.org/10.1074/jbc.M114.600569.
Kochlamazashvili, G., Henneberger, C., Bukalo, O., Dvoretskova, E., Senkov, O., Lievens, P.M.-J., Westenbroek, R., Engel, A.K., Catterall, W.A., Rusakov, D.A., et al. (2010). The Extracellular Matrix Molecule Hyaluronic Acid Regulates Hippocampal Synaptic Plasticity by Modulating Postsynaptic L-Type Ca2+ Channels. Neuron 67, 116–128. https://doi.org/10.1016/j.neuron.2010.05.030.
Perkins, K.L., Arranz, A.M., Yamaguchi, Y., and Hrabetova, S. (2017). Brain extracellular space, hyaluronan, and the prevention of epileptic seizures. Rev. Neurosci. 28, 869–892. https://doi.org/10.1515/revneuro-2017-0017.
Brakebusch, C., Seidenbecher, C.I., Asztely, F., Rauch, U., Matthies, H., Meyer, H., Krug, M., Böckers, T.M., Zhou, X., Kreutz, M.R., et al. (2002). Brevican-Deficient Mice Display Impaired Hippocampal CA1 Long-Term Potentiation but Show No Obvious Deficits in Learning and Memory. Mol. Cell. Biol. 22, 7417–7427. https://doi.org/10.1128/MCB.22.21.7417-7427.2002.
Favuzzi, E., Marques-Smith, A., Deogracias, R., Winterflood, C.M., Sánchez-Aguilera, A., Mantoan, L., Maeso, P., Fernandes, C., Ewers, H., and Rico, B. (2017). Activity-Dependent Gating of Parvalbumin Interneuron Function by the Perineuronal Net Protein Brevican. Neuron 95, 639-655.e10. https://doi.org/10.1016/j.neuron.2017.06.028.
Zhou, X.H., Brakebusch, C., Matthies, H., Oohashi, T., Hirsch, E., Moser, M., Krug, M., Seidenbecher, C.I., Boeckers, T.M., Rauch, U., et al. (2001). Neurocan is dispensable for brain development. Mol. Cell. Biol. 21, 5970–5978. https://doi.org/10.1128/MCB.21.17.5970-5978.2001.
Irala, D., Wang, S., Sakers, K., Nagendren, L., Ulloa Severino, F.P., Bindu, D.S., Savage, J.T., and Eroglu, C. (2024). Astrocyte-secreted neurocan controls inhibitory synapse formation and function. Neuron 112, 1657-1675.e10. https://doi.org/10.1016/j.neuron.2024.03.007.
Arranz, A.M., Perkins, K.L., Irie, F., Lewis, D.P., Hrabe, J., Xiao, F., Itano, N., Kimata, K., Hrabetova, S., and Yamaguchi, Y. (2014). Hyaluronan deficiency due to Has3 knock-out causes altered neuronal activity and seizures via reduction in brain extracellular space. J. Neurosci. Off. J. Soc. Neurosci. 34, 6164–6176. https://doi.org/10.1523/JNEUROSCI.3458-13.2014.
Carulli, D., Pizzorusso, T., Kwok, J.C.F., Putignano, E., Poli, A., Forostyak, S., Andrews, M.R., Deepa, S.S., Glant, T.T., and Fawcett, J.W. (2010). Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 133, 2331–2347. https://doi.org/10.1093/brain/awq145.
Bekku, Y., Saito, M., Moser, M., Fuchigami, M., Maehara, A., Nakayama, M., Kusachi, S., Ninomiya, Y., and Oohashi, T. (2012). Bral2 is indispensable for the proper localization of brevican and the structural integrity of the perineuronal net in the brainstem and cerebellum. J. Comp. Neurol. 520, 1721–1736. https://doi.org/10.1002/cne.23009.
Lam, D., Enright, H.A., Cadena, J., Peters, S.K.G., Sales, A.P., Osburn, J.J., Soscia, D.A., Kulp, K.S., Wheeler, E.K., and Fischer, N.O. (2019). Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci. Rep. 9, 4159. https://doi.org/10.1038/s41598-019-40128-1.
Vrselja, Z., Daniele, S.G., Silbereis, J., Talpo, F., Morozov, Y.M., Sousa, A.M.M., Tanaka, B.S., Skarica, M., Pletikos, M., Kaur, N., et al. (2019). Restoration of brain circulation and cellular functions hours post-mortem. Nature 568, 336–343. https://doi.org/10.1038/s41586-019-1099-1.
Lang, B.T., Cregg, J.M., DePaul, M.A., Tran, A.P., Xu, K., Dyck, S.M., Madalena, K.M., Brown, B.P., Weng, Y.-L., Li, S., et al. (2015). Modulation of the proteoglycan receptor PTPσ promotes recovery after spinal cord injury. Nature 518, 404–408. https://doi.org/10.1038/nature13974.
Yang, S., Hilton, S., Alves, J.N., Saksida, L.M., Bussey, T., Matthews, R.T., Kitagawa, H., Spillantini, M.G., Kwok, J.C.F., and Fawcett, J.W. (2017). Antibody recognizing 4-sulfated chondroitin sulfate proteoglycans restores memory in tauopathy-induced neurodegeneration. Neurobiol. Aging 59, 197–209. https://doi.org/10.1016/j.neurobiolaging.2017.08.002.
Keough, M.B., Rogers, J.A., Zhang, P., Jensen, S.K., Stephenson, E.L., Chen, T., Hurlbert, M.G., Lau, L.W., Rawji, K.S., Plemel, J.R., et al. (2016). An inhibitor of chondroitin sulfate proteoglycan synthesis promotes central nervous system remyelination. Nat. Commun. 7, 11312. https://doi.org/10.1038/ncomms11312.
Stephenson, E.L., Zhang, P., Ghorbani, S., Wang, A., Gu, J., Keough, M.B., Rawji, K.S., Silva, C., Yong, V.W., and Ling, C.-C. (2019). Targeting the Chondroitin Sulfate Proteoglycans: Evaluating Fluorinated Glucosamines and Xylosides in Screens Pertinent to Multiple Sclerosis. ACS Cent. Sci. 5, 1223–1234. https://doi.org/10.1021/acscentsci.9b00327.
Cathomas, F., Lin, H.-Y., Chan, K.L., Li, L., Parise, L.F., Alvarez, J., Durand-de Cuttoli, R., Aubry, A.V., Muhareb, S., Desland, F., et al. (2024). Circulating myeloid-derived MMP8 in stress susceptibility and depression. Nature 626, 1108–1115. https://doi.org/10.1038/s41586-023-07015-2.
Mai, Z., Lin, Y., Lin, P., Zhao, X., and Cui, L. (2024). Modulating extracellular matrix stiffness: a strategic approach to boost cancer immunotherapy. Cell Death Dis. 15, 1–16. https://doi.org/10.1038/s41419-024-06697-4.
Liu, C.-C., Zhao, N., Yamaguchi, Y., Cirrito, J.R., Kanekiyo, T., Holtzman, D.M., and Bu, G. (2016). Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-β clearance and aggregation in Alzheimer’s disease. Sci. Transl. Med. 8, 332ra44. https://doi.org/10.1126/scitranslmed.aad3650.
Howell, M.D., Bailey, L.A., Cozart, M.A., Gannon, B.M., and Gottschall, P.E. (2015). Hippocampal administration of chondroitinase ABC increases plaque-adjacent synaptic marker and diminishes amyloid burden in aged APPswe/PS1dE9 mice. Acta Neuropathol. Commun. 3, 54. https://doi.org/10.1186/s40478-015-0233-z.
Lopera, F., Marino, C., Chandrahas, A.S., O’Hare, M., Villalba-Moreno, N.D., Aguillon, D., Baena, A., Sanchez, J.S., Vila-Castelar, C., Ramirez Gomez, L., et al. (2023). Resilience to autosomal dominant Alzheimer’s disease in a Reelin-COLBOS heterozygous man. Nat. Med. 29, 1243–1252. https://doi.org/10.1038/s41591-023-02318-3.
Hiscox, L.V., Johnson, C.L., McGarry, M.D.J., Perrins, M., Littlejohn, A., van Beek, E.J.R., Roberts, N., and Starr, J.M. (2018). High-resolution magnetic resonance elastography reveals differences in subcortical gray matter viscoelasticity between young and healthy older adults. Neurobiol. Aging 65, 158–167. https://doi.org/10.1016/j.neurobiolaging.2018.01.010.
Knowles, T.P.J., and Buehler, M.J. (2011). Nanomechanics of functional and pathological amyloid materials. Nat. Nanotechnol. 6, 469–479. https://doi.org/10.1038/nnano.2011.102.
Ge, X., Xu, X., Cai, Q., Xiong, H., Xie, C., Hong, Y., Gao, X., Yao, Y., Bachoo, R., and Qin, Z. (2024). Live Mapping of the Brain Extracellular Matrix and Remodeling in Neurological Disorders. Small Methods 8, 2301117. https://doi.org/10.1002/smtd.202301117.
Benbenishty, A., Peled-Hajaj, S., Krishnaswamy, V.R., Har-Gil, H., Havusha-Laufer, S., Ruggiero, A., Slutsky, I., Blinder, P., and Sagi, I. (2023). Longitudinal in vivo imaging of perineuronal nets. Neurophotonics 10, 015008. https://doi.org/10.1117/1.NPh.10.1.015008.
Lemieux, S.P., Lev-Ram, V., Tsien, R.Y., and Ellisman, M.H. (2023). Perineuronal nets and the neuronal extracellular matrix can be imaged by genetically encoded labeling of HAPLN1 in vitro and in vivo. Preprint at bioRxiv, https://doi.org/10.1101/2023.11.29.569151 https://doi.org/10.1101/2023.11.29.569151.
Sterin, I., Niazi, A., Kim, J., Park, J., and Park, S. (2024). Novel extracellular matrix architecture on excitatory neurons revealed by HaloTag-HAPLN1. bioRxiv, 2024.03.29.587384. https://doi.org/10.1101/2024.03.29.587384.
 | A guest post by
|